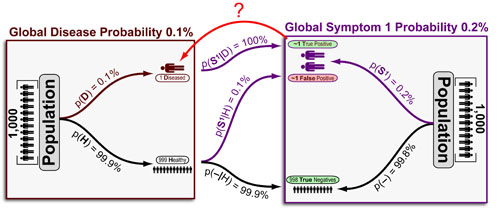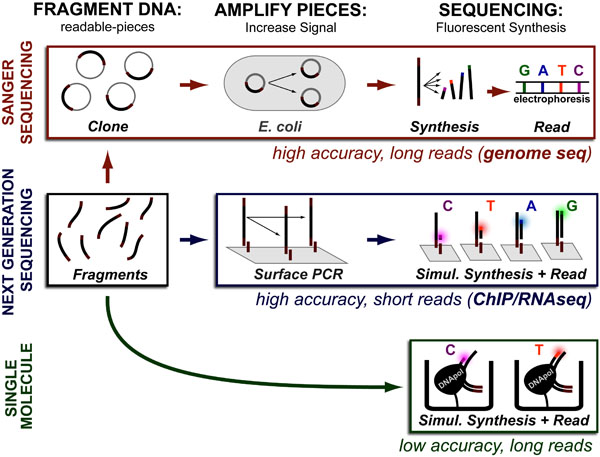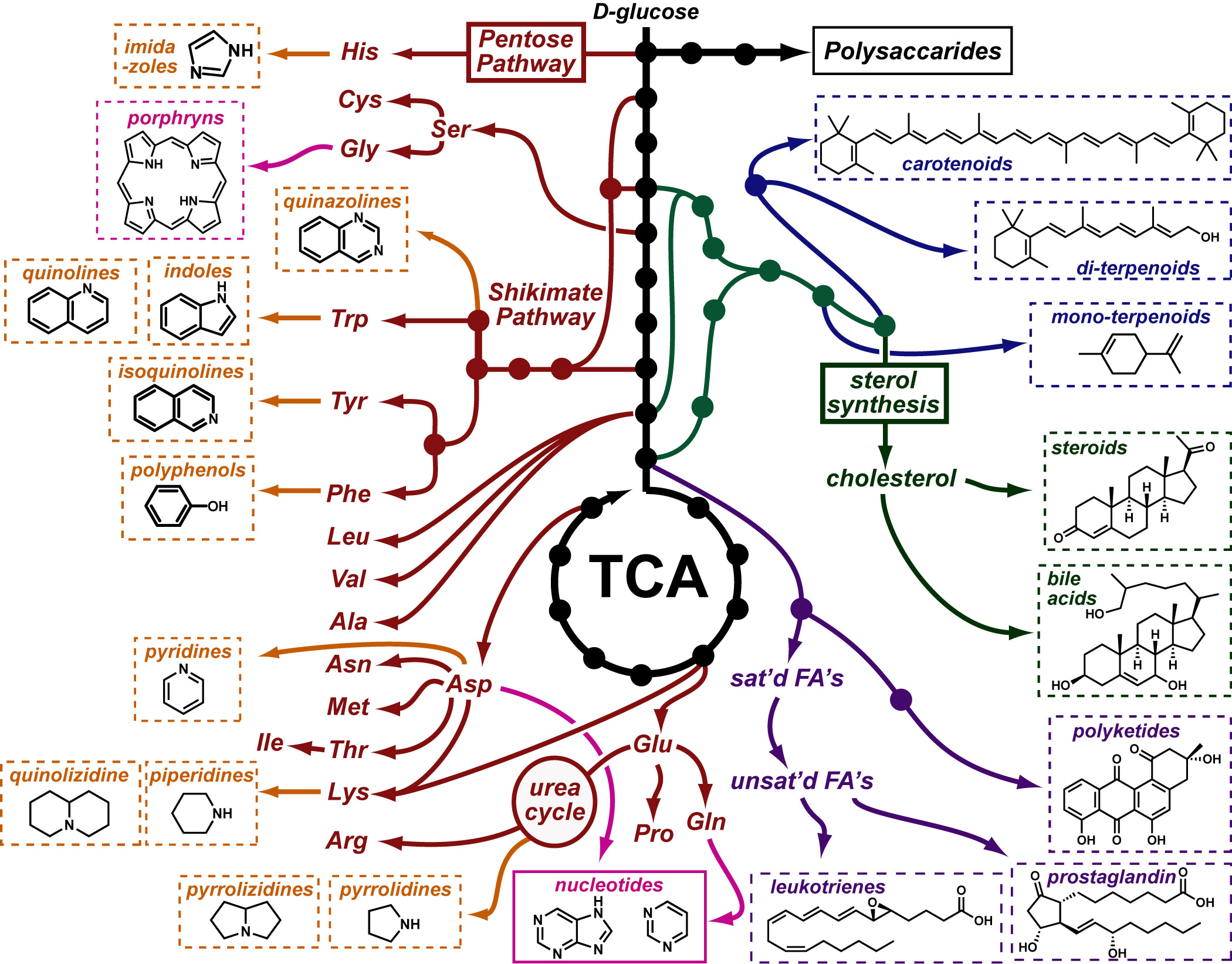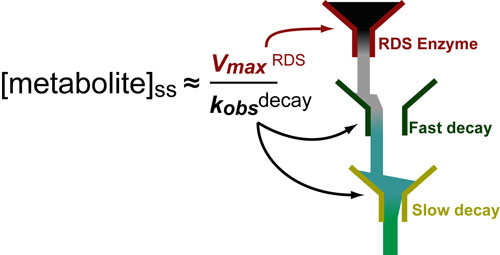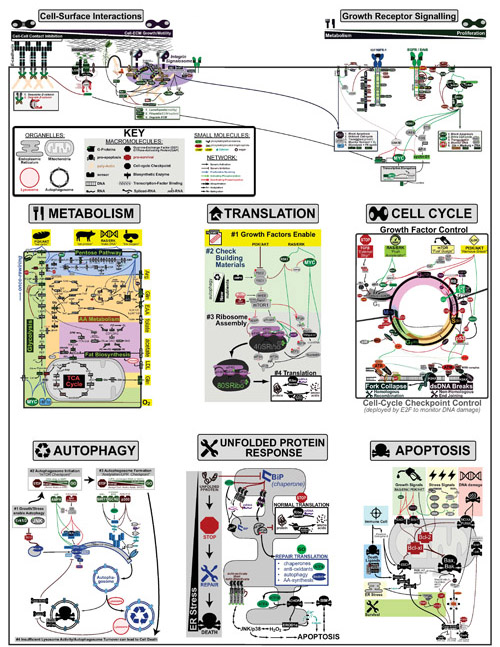
Cancer is a complex disease that is defined by at least 10 different “Hallmarks” that reflect mutation or epigenetically-driven reprogramming of normal cellular circuits. Over the past year, I have compiled a document that attempts to combine what’s known about the intra-cellular network that underlie these “Cancer Hallmarks.” This project started out as a single-page infographic but has since expanded into the 2 foot x 3 foot poster pictured above.
My goal was (and is) to create a comprehensive network map that is conceptually accessible to help me (and now others) think about the “big picture” of cancer networks. Of course, this poster is a work in progress and I will continue to update it over time. Below, I give a brief conceptual description of each module I have used to organize this “Canonical” Cancer-Network Map.